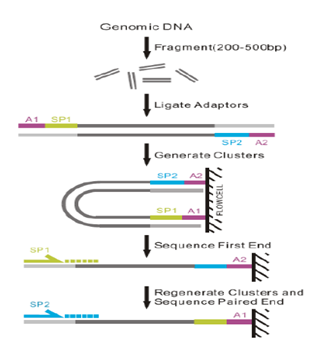
全基因组重测序(WGS)
- shbio
- 伯豪生物
- 上海市
- 现货
- 按需定制
- 议价
- 2022-09-16 08:31:26
上海伯豪生物技术有限公司
- 英文名称
- Whole Genome Sequencing,WGS
- CAS号
- -
- 别名
- 全基因组重测序,WGS
- 关键词
- 全基因组重测序WGS全基因组基因组重测序全基因组测序
- 科研服务
- 全基因组重测序(WGS):“是对已知基因组序列的物种的不同个体或群体进行全基因组重新测序,通过与原有基因进行比较,得到丰富的全基因组变异信息,并在个体或群体水平上进行生物信息分析。”“”

全基因组重测序(Whole Genome Sequencing,WGS)是对已知基因组序列的物种的不同个体或群体进行全基因组重新测序,通过与原有基因进行比较,得到丰富的全基因组变异信息,并在个体或群体水平上进行生物信息分析。


Illumina 高通量测序平台

全基因组重测序可以检测单核苷酸多态性(SNP)、插入缺失(InDel)、拷贝数变异(CNV)、染色体结构变异(SV)等,发现 DNA 变异与某一表型(例如某种疾病)之间的联系。由于不需要重新从头拼接,因此仅需要相对较小的数据量,通过基因组比对(Mapping)的方式得到个体序列变化的信息。随着测序成本的降低和分析技术的成熟,目前全基因组重测序在人类和动植物遗传学研究中已有广泛应用。

- 十余年样本处理经验;
- 丰富的易降解样本处理经验;
- 多层样本质量控制,保证数据质量;
- 丰富的样本处理经验,涉及组织、细胞、全血、血清、血浆、FFPE 等各类实验样本。
- 涵盖肿瘤、遗传病、复杂疾病、遗传育种等各个领域,协助客户在 Nature、Nat Genet、Nat Med、J Clin Oncol、Leukemia、PNAS、Cell Res 等高水平期刊发表论文 100+ 篇(截止 2019 年 6 月,伯豪生物协助客户发表SCI文章数)。


- 样本类型:DNA、组织、细胞、全血等;
- DNA 总量:DNA≥ 2 μg;浓度:DNA≥ 50 ng/μl;要求主带清晰,无降解,浓度大于 50ng/ul,总量大于 1ug,OD260/OD280 =1.8~2.0,OD260/OD230=1.5 ~2.2。
- 测序深度:通常≥30X,视具体项目需求而定;
- 测序读长:2*150bp。

案例一:全基因组关联分析揭示大豆农艺性状的遗传网络

不同复杂性状间的耦合是分子设计育种的关键科学问题。作物的产量、品质等大都是多基因控制的复杂性状,由于受到一因多效和遗传连锁累赘的影响,使某些性状在不同材料和育种后代中协同变化,呈现耦合性相关。解析大豆复杂性状间耦合的遗传调控网络,明确关键调控单元,对于高产优质大豆新品种的培育具有重要意义。
材料和方法:对 809 份大豆栽培材料进行了重测序(平均深度为 8.3×),并对其遗传多样性进行了分析,明确了这些材料的群体结构。进而,对这 809 份材料的 84 个产量和品质性状进行了连续多年多点的观测。进一步,该团队利用全基因组关联分析并结合新开发的上位性效应检测方法,对 84 个性状的调控位点进行了系统的全基因组扫描,解析不同农艺性状之间的内在遗传调控网络。

1、鉴定出 84 个性状的 245 个显著性关联位点
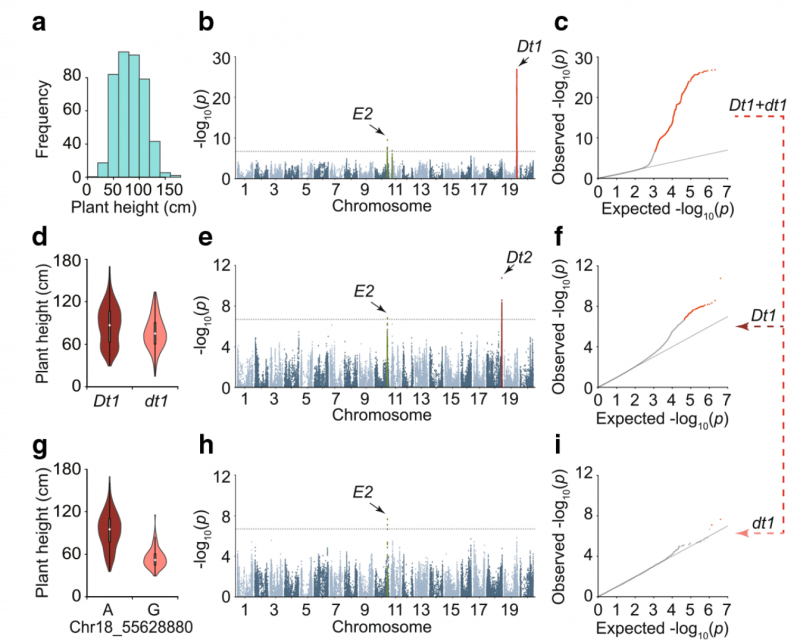
▲图 1:大豆株高的 GWAS。【a】809 个大豆品种株高的分布;【b】 所有品种 GWAS 的结果,在 GWAS 结果中,已知的基因 DT1 和 E2 都被鉴定出来;【c】株高的 Q - Q 图;【d】DT1 等位基因分组后株高的变化。已知的基因 DT1 将 809 份亲本分为两个亚组,具有不同的株高平均值;【e】 使用 DT1 亚组进行 GWAS 的结果;【f】DT1 亚组进行 GWAS 的 Q - Q 图;【g】DT1 亚群内不同 DT2 基因型分组后的株高变异;【h】 使用 dt1 亚组进行 GWAS 的结果;【i】dt1 亚组进行 GWAS 的 Q - Q 图;水平虚线表示 GWAS 的显著阈值(2 × 10–7)。
2、脂肪酸和脂代谢相关的基因
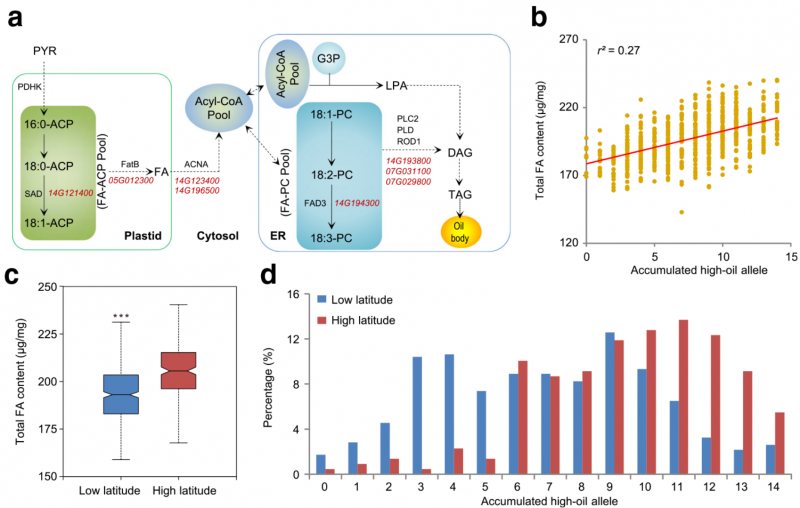
▲图 2:大豆脂肪酸含量遗传调控的初步探讨。【a】候选基因参与脂肪代谢途径,主要参与大豆种子中脂肪酸(FA)合成的变异;虚线代表多个反应步骤;【b】总脂肪含量与高油含量等位基因数目的关系图;【c】低纬度和高纬度地区种质的总 FA 含量;【d】低纬度和高纬度群体中高油等位基因的比例。
对于油含量相关性状,共鉴定到 24 个脂肪酸代谢相关和 21 个脂代谢相关的基因,它们分别参与了不同的重要酶促反应。深入分析发现这些基因是通过加性效应共同调控多个大豆油脂性状的形成。
3、大豆不同性状间的遗传调控网络
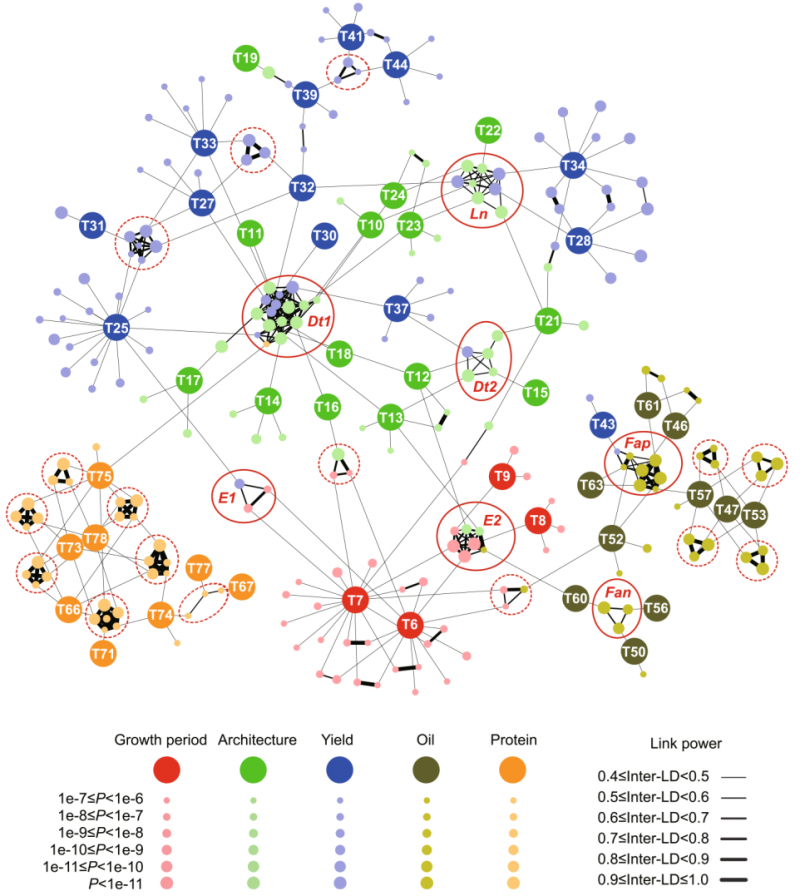
图 3:大豆不同性状间的遗传调控网络;节点表示性状及其对应的关联位点;不同性状关联位点之间的边由 LD 决定;涉及 Dt1,Dt2,E1,E2,Ln,Fan 和 Fap 的位点用实线圈表示,未知的位点用虚线圈表示。
这些关联位点揭示了不同性状间相互耦合的遗传基础。根据连锁不平衡分析,发现 115 个关联位点可相互连锁,并将所观测的 51 个性状联系起来,形成复杂的多性状多位点调控网络,该遗传调控网络很好地解释了不同性状间的耦合关系。研究还发现其中 23 个关联位点起到了重要节点作用,对不同性状的形成起到关键调控作用,并对其中部分位点在不同性状耦合中的作用进行了验证。

Fang C , Ma Y , Wu S , et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean[J]. Genome Biology, 2017, 18(1):161. (IF=14.028).