Nature | 迄今为止最大规模GWAS研究绘制身高基因图谱,揭示1.2万个与人类身高相关的SNP
在婴儿成长为成人过程中,遗传因素起到了重要作用,这一直是人类生物学中一个复杂而又鲜为人知的领域。自2007年以来,全基因组关联研究(GWAS)已经确定了常见单核苷酸多态性(SNP)与身高之间的数千种关联。此前最大规模的、有关身高的GWAS纳入了多达70万个体的样本量,并报告了712个位点的3,290个独立关联。但这些遗传数据大部分来自欧洲血统人群,得出的结果无法涵盖到全球范围。在欧洲血统以外的人群
近日,由昆士兰大学的科学家领导、全球多个研究单位和机构的600余名研究人员参与的基因组学研究成果以“A saturated map of common genetic variants associated with human height”为题在国际顶尖期刊Nature发表。研究团队分析了来自281个GWAS、近540万人的遗传数据,其中超过100万的参与者是非欧非裔、东亚裔、西班牙裔或南亚裔,克服了早期进行的小型研究中数据分散的局限性。
该研究首次确定了基因组中身高相关变异集群,提供了包含12,111个与身高相关常见变异的特定基因组区域的综合图谱,揭示身高与遗传力和多个特定的基因组区域有关,填补了人们在理解基因差异如何导致身高差异方面的巨大空白。
文章发表在Nature
主要研究内容
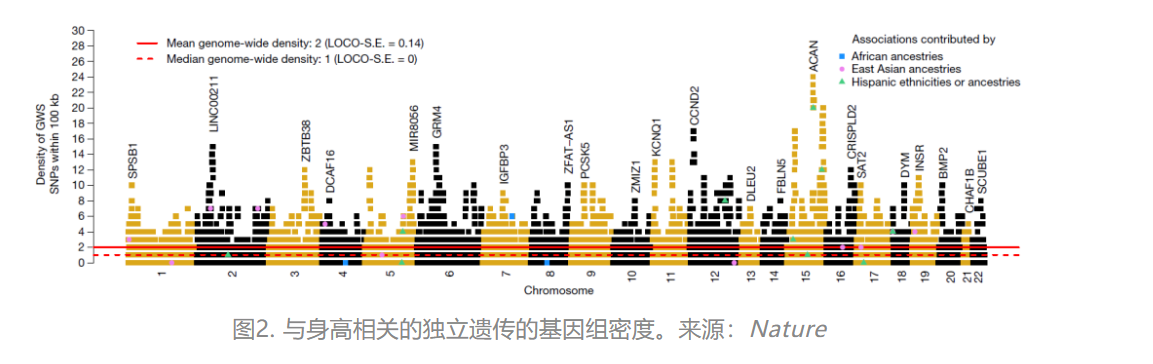
系统性分析共确定12,111个与身高相关的SNP
研究团队对来自GIANT联盟项目和23andMe公司的281个研究中5,380,080名个体进行了遗传分析,其中包括4,080,687名欧洲血统参与者(占总样本的75.8%,EUR)、472,730名东亚血统参与者(8.8%,EAS)、455,180名典型混合血统的西班牙裔参与者(8.5%,HIS)、293,593名以非洲血统为主的参与者(5.5%,AFR)和77,890名主要来自南亚血统的参与者(1.4%,AFR)。
研究团队在五组队列中分别进行了meta分析,并专门研究了所有祖先特异性GWAS中的混杂效应,以使分析结果受种群分层的影响最小。为比较五组队列的分析结果,研究团队检测了在EUR队列中确定的SNP与在其他队列中发现的SNP之间的遗传和物理共定位,发现身高相关Genome-Wide Significant(GWS)SNP在主要祖先群体中基本相同。
图1. 等位基因频率与估计效应大小之间的关系。来源:Nature
为找到特定群体的信号,研究团队测试了在非EUR GWAS中检测到的单个SNP是否与在EUR GWAS中检测到的信号有条件地独立。使用特定于每个祖先群体的连锁不平衡结构(LD)参考panel拟合了一个GWS SNP的近似联合模型,在排除强LD中的SNP后,研究团队在4个非EUR队列中分别检测到2、17、49和63个有条件地独立于EUR GWAS中确定的GWS SNP。
接下来,研究团队对上述meta分析结果进行COJO分析,从多血统meta分析中鉴定出半独立的GWS SNP,并使用来自英国生物银行(UKB)中约 350,000 名无关EUR参与者的基因型作为LD参考,最终确定了12,111个准独立的GWS SNP,包括9,920个具有GWS边际效应的主信号和2,191个在联合回归模型中仅达到GWS的次信号,其主要集中在与机体骨骼生长相关的基因组区域。
由已识别位点内SNP解释身高差异
研究团队将12,111个GWS SNP分为7,209个不重叠的位点,其长度从70kb(仅包含一个信号的位点)到711kb(包含多达25个信号的位点)不等。GWS位点的平均长度约为90kb(平均值为46kb),累积长度约为647Mb,约占基因组的21%。
为估计由基因组中21%的GWS重叠位点内的共同变异所决定的遗传力比例,研究团队计算了两个基因组关系矩阵(GRMs),并使用这两个矩阵来估算5个队列中基于SNP的身高分层遗传力。结果显示,基于SNP的身高分层遗传力可解释约100%的欧洲人群和超90%非欧人群的身高分析结果。上述结果表明,几乎所有可归因于常见遗传变异的身高变异都可以映射到约21%的基因组区域。
图3. GWS位点内SNP解释了身高的差异。来源:Nature
结语
综上所述,该研究对540万人进行了迄今为止规模最大的GWAS研究,为身高的遗传结构提供了深入见解,并绘制了包含12,111个身高相关遗传关联的基因组图谱。该研究结果能够帮助医生识别那些无法达到基因预测身高的人群,从而帮助诊断可能阻碍其生长或影响其身体健康的隐藏疾病或状况。此外,该研究还为通过全基因组研究来确定导致疾病和相关生物表型的遗传成分提供了研究框架。
伦敦大学学院遗传流行病学教授Karoline Kuchenbaecker在同期Nature杂志上发表了一篇关于该研究的独立观点文章,指出:“这一规模化研究标志着我们理解遗传对复杂性状贡献的一个里程碑,也凸显了拟合现有基因数据多样性差距的重要工作仍有待完成。”
文章发表在Nature
文章通讯作者&第一作者Loïc Yengo博士表示:“人与人之间80%的身高差异是由遗传因素决定的。我们发现的12000个变异解释了40%的身高差异,这意味着我们为利用DNA来预测身高打开了大门。”
文章共同第一作者、伦敦玛丽女王大学计算生物学高级讲师Eirini Marouli博士表示:“基因组研究是革命性的,可能是解决许多全球健康挑战的关键。如果我们能将基因组的特定区域映射到某些特征上,可为未来广泛的、有针对性的、个性化的治疗打开大门,这将造福世界各地的人们”。
参考文献:
1. Loïc Yengo, A saturated map of common genetic variants associated with human height, Nature (2022).
2. Márquez-Luna, C. et al. Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets. Nat. Commun. 12, 6052 (2021).
3. Halldorsson, B. V. et al. The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740 (2022).

咨询
我们尊重知识产权,如您认为本平台所载文章、图片、视频等内容侵犯您的合法权益,请您及时联系我们,我们将依据相关法律法规、平台规则予以处理。
关键字
- 194
- 点赞
- 复制链接
- 举报