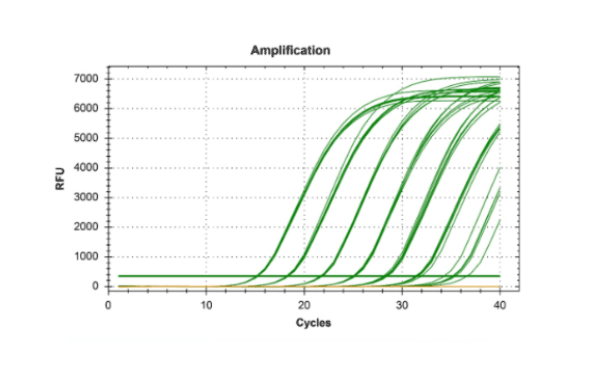
qPCR体系优化和常见问题分析
qPCR实验的工作流程首先需要确定研究的目的,根据实验设计规划好实验分组、重复次数等细节。接下来分为样本准备和引物探针验证两个重要的步骤。样本准备主要是核酸提取逆转录等步骤,引物探针需要去测试特异性和效率。接下来需要使用qPCR仪来对样品中的目的核酸进行扩增qPCR结束后根据实验目的对目的核酸进行相对或者绝对定量。
前言
聚合酶链式反应(PCR)是用于扩增特定DNA片段的分子生物学实验技术。实时荧光定量PCR(以下简称qPCR)作为第二代PCR技术,自1996年推出以来,已经广泛应用于基因表达分析、病原微生物检测、动植物育种等许多研究领域,为了获得最理想的检测结果,qPCR从样本采集、核酸提取、cDNA合成到上机检测的流程有许多可以优化的参数。
qPCR实验的工作流程首先需要确定研究的目的,根据实验设计规划好实验分组、重复次数等细节。接下来分为样本准备和引物探针验证两个重要的步骤。样本准备主要是核酸提取逆转录等步骤,引物探针需要去测试特异性和效率。接下来需要使用qPCR仪来对样品中的目的核酸进行扩增qPCR结束后根据实验目的对目的核酸进行相对或者绝对定量。接下来讲的qPCR体系优化会围绕着这个流程展开。
1.样本的采集与处理
首先,提前做好功课,了解样本的不同分型,或者了解详细的细胞分群。如果条件允许尽可能覆盖所有的组织类型或者细胞类型。其次,尽可能增加样本数量,也就是生物学重复,从而更客观地反映生物变异程度。另外,qPCR实验也需要有技术重复来降低误差。
采样是需要严格规划的过程,比如材料的时效性、珍贵程度等,都要纳入考量范围。样品要尽量新鲜,取样尽可能快速。戴手套操作,防止污染。如果不马上提取核酸,需要-80°C保存,并尽快处理。

2.核酸的提取和检测
模板的质量直接影响到检测性能。核酸提取需要有效地将RNA或DNA从其他混合物中分离。RNA样本中的污染物——基因组DNA、DNA结合蛋白、酚类化合物或在提取RNA过程中引入的外源杂质(如手套中的粉末)——都已被证明会抑制下游实验,如逆转录和PCR扩增。核酸提取需要使用无菌无酶的试剂耗材,避免RNase或DNase污染,并对内源RNAse或DNAse进行有效抑制;多糖多酚样品要考虑多糖多酚杂质的有效去除。低温保存防止RNA或DNA降解。
降解或不纯的RNA会限制逆转录反应的效率,降低产量。部分降解的RNA可能不能给出准确的基因表达结果。对于基因的定量,必须使用高质量的RNA,这意味着需要非常仔细地检查RNA的浓度和质量。可采用高分辨率琼脂糖凝胶检测核酸质量和分光光度法(A260/A280=1.8和A260/A230=2.0)检测核酸纯度和浓度。
3.cDNA合成
RNA 质量对 cDNA 合成结果会产生重要影响。并且RNA 很脆弱,容易降解。为了保证 RNA 的完整性,我们需要非常注意,比如在冰上操作,用 RNase-free 的枪头和离心管,减少操作时间等。在反应体系中加入 RNase 抑制剂也能有效防止 RNA 降解。
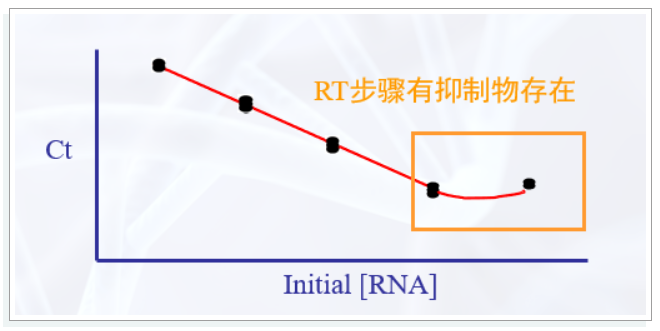
如何评价样品中的杂质对逆转录的影响呢?可以梯度稀释后绘制标准曲线,如果低浓度的样品点数值偏大比较明显,基本可以判定杂质影响显著。
不同厂家的反转录试剂会有差异,对RNA中的杂质耐受程度也不同。逆转录酶在整个反转录体系中具有关键性影响。除了活性以外,逆转录酶的热稳定性同样很重要,在较高温度下进行逆转录,能够减少 RNA 的二级结构,增加逆转录的效率。
除了掌握 RNA 的完整性之外,反转录之前还需要对 RNA 浓度进行测定。一般反转录试剂盒会对上样量有要求,建议 total RNA 上样量小于 5 μg。超过这个范围,会使反转录产物产生偏好性 (表达丰度高的基因优先被反转录) 而造成定量结果不准确。
逆转录出来的cDNA可以直接放在4°C保存,若长期不用,可分装,然后-20°C保存。
4.qPCR方法的建立
① 定量方法
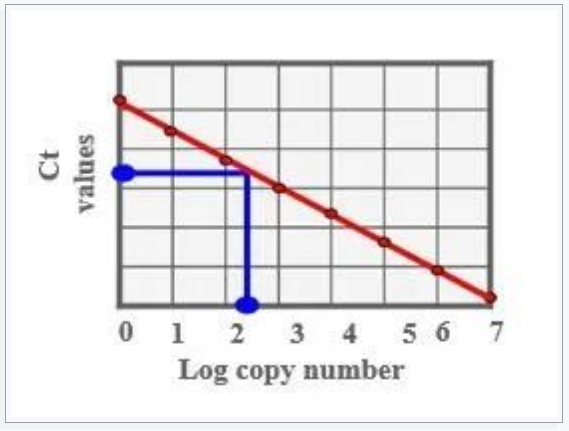
绝对定量:检测起始模板数的精确拷贝数,需要标准品构建标准曲线。
标准品可以是纯化的基因组DNA、质粒DNA或者体外转录RNA(cDNA),其作用是生成标准曲线,建立Ct值与浓度之间的线性关系。
标准品与待测样品的PCR效率一致,且接近100%,与样品的性质尽可能接近,与样品相同的扩增条件(PCR体系、耗材、同一次扩增),大于或等于5个梯度稀释的标准品。
相对定量:在一个样本中,目的基因相对于内参基因的量的变化。
内参基因选择建议筛选不少于三个内参基因来归一化RT-qPCR数据。目的是消除外部样品偏差,例如总RNA含量,RNA稳定性,酶效率或样品装载量的变化。
对候选的内参基因进行qPCR 实验,得出Ct平均值以及 Ct值的标准偏差,选择SD最小的基因作为实验内参。可通过geNorm 、 BestKeeper 、 NormFinder、RefGenes 等工具来评估您的内参基因。
② 荧光标记方法
染料法:利用能与DNA双链结合的染料来实现,如SYBR Green I。该染料在游离状态下呈现微弱的荧光,一旦与双链DNA的双螺旋小沟结合,其绿色荧光增强约1000倍。因此其总的荧光强度与双链DNA含量成正比,利用这一关系可以反映生成的PCR产物的量。
TaqMan荧光探针:是一种寡核苷酸探针,荧光基团连接在探针的5'末端,而淬灭剂则在3'末端。PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,探针完整时,报告基团发射的荧光信号被淬灭基团吸收; PCR扩增时, Tag酶的5'-3'外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。常用的荧光基团是FAM,TET,VIC,HEX。
引物探针设计可以参考Gene π网站:https://www.gene-pi.com/item/primers-and-probes-2/
③ 引物扩增效率验证
标准曲线是评估PCR扩增效率最可靠和稳定的一种方法,该方法涉及到制作一系列的样品来控制目标模板的相对数量。最常用的是10倍梯度稀释样品,采用标准qPCR程序进行扩增获得Cq值,最后根据各样品浓度及相应的Cq值绘制标准曲线,得到线性方程Cq= -klgX0+b,扩增效率E=10(-1/k)-1。利用qPCR进行定量分析时,要求扩增效率范围在90%-110%(3.6>k>3.1)。
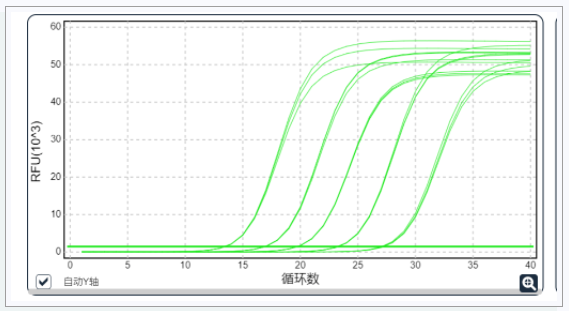
④ 反应体系优化
▶ 根据仪器类型,选择合适的耗材和qPCR试剂。
▶ 每对引物先进行预实验,确定特异性以及最适浓度。
▶ 配置不同的PCR反应体系,选择每个组分合适的浓度。
▶ 设置温度梯度测试引物最合适的退火温度。
▶ 实验设置NTC、NRT、 NEG和POS等对照组,来监控实验体系或污染。
实时荧光定量PCR常见问题分析
1.可疑的扩增曲线
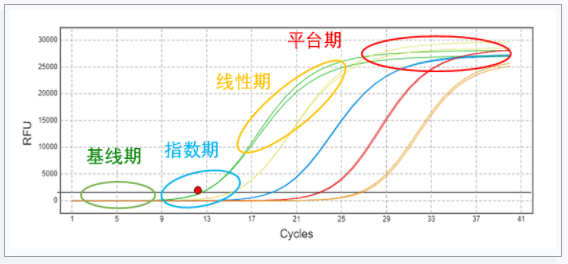
真正的扩增曲线,有特征的形状:首先背景信号,然后是三个增长阶段(指数增长期、线性增长期和平台期)。
如果不是同时具有特征性的三个增长阶段,没有典型的指数增长期,那就不存在扩增。
平台期很低也是常见的异常扩增曲线。可能是模板的浓度太低。通常如果模板的起始浓度太低, 反应体系中会形成大量的引物二聚体。大量引物二聚体的形成使得引物很快消耗完,从而造成扩增曲线的平台期很低。这种情况可通过调整引物和模板的比例。
2.异常的荧光信号
NTC出现荧光信号---引物二聚体形成或气溶胶污染,查看熔解曲线是否为单一峰。
3.扩增效率过高或过低
过低的扩增效率(<90%)可能存在的原因:
▶ 移液器校准不良或移液技术差。
▶ 不正确的稀释导致标准曲线出现错误。
▶ 引物设计不好或扩增子具有二级结构。
▶ 标准曲线动态范围太小。
▶ Taq酶无活性或活性降低。
▶ 样品抑制。
过高的扩增效率(> 110%)可能存在的原因:
▶ 移液器校准不良或移液技术差。
▶ 不正确的稀释导致标准曲线出现错误。
▶ 引物二聚体或非特异性扩增。
▶ 标准曲线动态范围太小。
▶ 基因组DNA污染。
4.重复性差
为精确定量,对每个样品都要做重复实验,复孔之间的Ct值不应超过0.5,标准偏差不大于0.2,这样,实验结果就有很好的精确度。
造成重复性差的原因:
▶ 加样误差(操作或者加样器导致)。
▶ 没有将试剂和样品充分混匀。
▶ 低拷贝的目的片段→泊松分布。
▶ 基线阈值设定不合理。
Cielo™实时荧光定量PCR系统
Harness of the power of qPCR

☑ 数据可靠性:连续1000次实验后,结果高度一致。
☑ 应用灵活性:提供多种qPCR应用分析。
☑ 流程智能化:中英文用户界面,触控操作,可多机联用。
☑ 在线便捷性:主机可独立运行qPCR程序,数据可USB、Wi-Fi等网络传输。
咨询
- 872
- 点赞
- 复制链接
- 举报