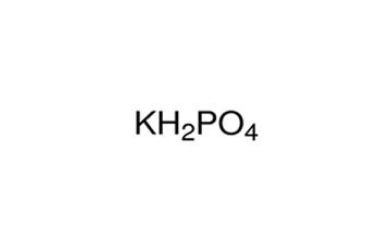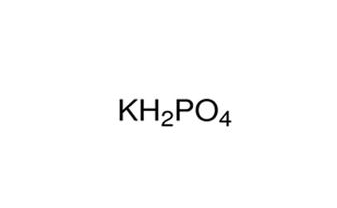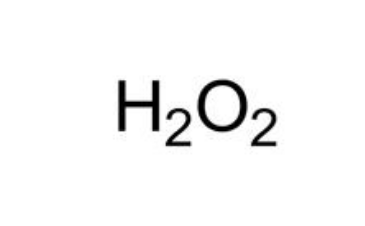

代谢组学研究中使用的高通量方法会产生大量代谢分析相关数据,这就需要采用生物信息学的方法进行处理。百泰派克公司生物信息学分析人员能够对代谢组学数据知识进行深度挖掘和全面分析,从质谱原始数据出发,进行峰对齐、保留时间校正和峰面积提取。代谢物结构鉴定采用精确质量数匹配(<25 ppm)和二级谱图匹配的方式,检索METLIN数据库和HMDB数据库;两组比较时,删除两组组内缺失值均>50%的离子峰;接着对数据进行归一化(采用autoscaling或UV法)。百泰派克应用MetaboAnalysis、SIMCA-P软件进行多维统计分析和单维统计分析,包括PCA、PLS-DA、OPLS-DA、以及通路富集分析,确保提供全面精准的代谢组学分析结果。
代谢组学生物信息学分析

代谢组学数据质量评估
百泰派克采用QC样本谱图比对和主成分分析两种方法,项目实验中的QC样本数据进行分析评价。将分析得到的QC样本UPLC-QTOF-MS总离子流图,进行谱图重叠比较,见下图。结果表明各色谱峰的响应强度和保留时间基本重叠,说明在整个实验过程中仪器误差引起的变异较小。

QC样品在正离子模式下TIC重叠图

主成分分析(PCA)
主成分分析(Principal Component Analysis, PCA)是将原本鉴定到的所有代谢物重新线性组合,形成一组新的综合变量,同时根据所分析的问题从中选取2-3个综合变量,使它们尽可能多地反映原有变量的信息,从而达到降维的目的。同时,对代谢物进行主成分分析还能从总体上反应组间和组内的变异度。总体样本PCA分析采用PCA的方法观察所有各组样本之间的总体分布趋势,找出可能存在的离散样本,综合考虑各种因素(样品数,样品珍贵程度,离散程度)决定离散点的除去与否。所有样本PCA得分图见下图(对样本进行两两分析的PCA得分图)。
-主成分分析得分图")
图1 主成分分析得分图
百泰派克采用XCMS软件对代谢物离子峰进行提取。将25个实验样本和QC样本提取得到的峰,归一化后进行PCA分析,如图所示QC样本(黑色) 紧密聚集在一起,表明本次试验的仪器分析系统稳定性较好,试验数据稳定可靠,在试验中获得的代谢谱差异能反映样本间自身的生物学差异。
-总样品的PCA得分图")
图2 总样品的PCA得分图

PLS-DA/OPLS-DA二维图
不同于主成分分析(PCA)法,Partial Least Squares Discrimination Analysis(PLS-DA)或Orthogonal PLS-DA(OPLS-DA)是一种有监督的判别分析统计方法。该方法运用PLS-DA建立代谢物表达量与样品类别之间的关系模型,来实现对样品类别的预测。分别建立两两分组比较的PLS-DA模型(图1)或OPLS-DA模型(图2),模型得到的参数评价会以表格形式提供。其中R^2X和R^2Y分别表示所建模型对X和Y矩阵的解释率,Q2标示模型的预测能力,理论上R^2、Q^2数值越接近1说明模型越好,越低说明模型的拟合准确性越差,通常情况下,R^2、Q^2高于0.5(50%)较好,高于0.4即可接受,且两者差值不应过大。临床样本由于个体差异大,不可控,尤其大样本时,R^2、Q^2大小为0.2左右亦可。图3则是对PLS-DA模型(c)的检验,直线的斜率大,Q^2的截距为X,说明PLS-DA模型没有过拟合。同时通过计算变量投影重要度(Variable Importance for the Projection, VIP)来衡量各代谢物的表达模式对各组样本分类判别的影响强度和解释能力,从而辅助标志代谢物的筛选(通常以VIP值>1.0作为筛选标准)(图4)。
-假手术组和模型组的PLS-DA得分图")
图1 假手术组和模型组的PLS-DA 得分图
-假手术组和模型组的OPLS-DA模型")
图2 假手术组和模型组的OPLS-DA模型
-PLS-DA模型的排列检验图")
图 3 PLS-DA模型的排列检验图
-假手术组和模型组的PLS-DA模型载荷图")
图 4 假手术组和模型组的PLS-DA 模型载荷图
注:红框所圈的点为VIP>1的代谢产物

数据归一化分析
数据的完整性和准确性是后续获得具有统计学和生物学意义的分析结果的必要条件。在确保实验设计的合理性和实验数据的准确性的基础上,百泰派克首先对数据的完整性进行检查,对缺失值进行删除或者补充,删除极值,并对数据进行样本间和代谢物间的归一化处理,以确保各样本之间和代谢物之间可平行比较。原始数据中,缺失值超过50%的代谢物将被去除,不参与后续分析;对代谢物的表达量进行对数转换并利用Autoscaling方法(Mean-centered and divided by the standard deviation of each variable)进行归一化处理。下图显示了正离子模式数据经归一化处理前后的分布情况,结果表明数据经归一化处理后基本呈正态分布。

归一化前后的样本

单变量统计分析
在进行两组样本间的差异代谢物分析时,常用的单变量分析方法包括变异倍数分析(Fold Change Analysis, FC Analysis)、T检验,以及综合前两种分析方法的火山图(Volcano Plot)。利用单变量分析可以直观地显示两样本间代谢物变化的显著性,从而帮助我们筛选潜在的标志代谢物(通常以FC>2.0 且 P value<0.05作为筛选标准)。下图显示了数据的火山图,图中枚红色点为FC>2.0且P value<0.05的代谢物,即单变量统计分析筛选的差异代谢物。其余各组的火山图会以附件形式给出。

火山图分析
注:数据结果的火山图(Volcano Plot),绿、红色点为显著性差异代谢物(P value<0.05) 百泰派克选择有多维统计分析筛选标准(VIP>1)和单变量统计分析筛选标准(FC > 2.0且P value<0.05)的代谢物作为具有显著性差异的代谢物(图12)。鉴定出的显著性差异代谢物会以表格形式给出。

显著性差异的代谢物

差异代谢产物聚类分析
为了评价候选代谢物的合理性,同时更全面直观地显示样本之间的关系以及代谢物在不同样本中的表达模式差异,我们利用定性的显著性差异代谢物的表达量对各组样本进行层次聚类(Hierarchical Clustering),从而辅助我们准确地筛选标志代谢物,并对相关代谢过程的改变进行研究。一般来说,当筛选的候选代谢物合理且准确时,同组样本能够通过聚类出现在同一簇( Cluster)中。同时,聚在同一簇内的代谢物具有相似的表达模式,可能在代谢过程中处于较为接近的反应步骤中。下图显示了显著性差异代谢物层次聚类结果。

图 1显著性差异代谢物层次聚类结果

KEGG差异代谢产物通路分析
百泰派克将得到的差异代谢物使用MBRole进行代谢通路富集,使用KEGG数据库作为背景,进行相关通路分析。选取同物种的所有代谢物作为背景,分析 P value<0.05的代谢通路。图1和2为我们可提供的两种代谢通路图,富集的代谢通路以及相关T检验分析结果会以表格的形式给出。

图1 差异代谢物通路图

图2 代谢物通路图